
Introduction
In the field of medical research, harnessing the power of advanced technologies has become paramount in unlocking new insights and accelerating discoveries. One such innovation, the Forward-Looking Active Retrieval Augmented Generation framework, offers a groundbreaking approach to information retrieval and generation tasks. In this blog post, we’ll delve into the capabilities of FLARE when integrated with a Knowledge Graph and the Llama 3 8B language model, a cutting-edge sparse mixture of experts architecture renowned for its prowess in natural language understanding. Leveraging the LlamaIndex framework we’ll explore the transformative potential of FLARE in the domain of cancer research datasets.
With FLARE, researchers can dynamically retrieve relevant information from vast knowledge repositories while generating insightful analyses and summaries in real time. By incorporating a Knowledge Graph, FLARE gains access to structured domain-specific information, which enables more contextually relevant and accurate generation outputs. The Llama 3 8B language model, renowned for its robustness and sophistication, further enhances FLARE’s capabilities, by enabling nuanced understanding and interpretation of complex medical data.
Through this comprehensive guide, researchers and practitioners in the field of oncology can gain a deeper understanding of FLARE’s capabilities and its potential impact on accelerating cancer research. Before we get started, let’s take a look at Forward-Looking Active RAG (FLARE).
An Overview of Forward-Looking Active RAG
What Is Active RAG?
Active RAG stands for Active Retrieval Augmented Generation. It’s a framework proposed for enhancing long-form text generation models, particularly large language models, with the ability to actively retrieve relevant information from external sources during the generation process. The goal of Active RAG is to improve the quality, relevance, and factuality of the generated text by dynamically fetching additional knowledge when needed.
The Active RAG framework involves iteratively generating text while simultaneously assessing the need for additional information retrieval. If the model encounters uncertainty or lacks relevant knowledge during the generation process, it triggers a retrieval mechanism to fetch relevant information from external sources. This retrieved information is then incorporated into the ongoing generation process to produce more informed and contextually relevant output.
The Concept of FLARE
FLARE, which stands for Forward-Looking Active Retrieval Augmented Generation, is a specific implementation of the Active RAG framework. FLARE introduces a method for language models to actively retrieve relevant information from external sources while generating text. The concept of FLARE revolves around the idea of forward-looking retrieval, where the LLM anticipates its future information needs during the generation process. It achieves this by iteratively assessing the content it is about to generate and determining if there are gaps in its knowledge or uncertainties that require additional information.

FLARE offers two main methods for active retrieval:
- FLARE with Retrieval Instructions: In this method, the LLM generates retrieval queries using retrieval-encouraging instructions. These instructions prompt the LLM to retrieve information when necessary while generating the answer.
- Direct FLARE: This method involves using the LLM’s generation as search queries. The LLM iteratively generates the next sentence and retrieves relevant documents if uncertain tokens are present, regenerating the next sentence based on the retrieved information.
Working of FLARE with Retrieval Instructions
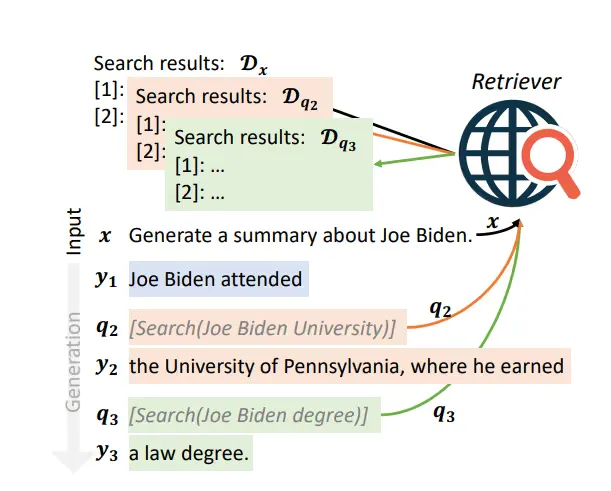
Here’s how FLARE with retrieval instructions works:
- Retrieval Instructions: In FLARE with retrieval instructions, the LLM is provided with explicit prompts or instructions to guide it in generating search queries. These prompts indicate when additional information is needed and instruct the LLM to initiate retrieval accordingly.
- Prompt Structure: The retrieval instructions are structured in a way that combines two skills: one for guiding LLMs to generate search queries and another for performing a specific downstream task, such as question-answering or summarization. The LLM is trained to combine these skills during inference to generate search queries while performing the task.
- Integration with Downstream Tasks: During inference, the LLM receives a test case that requires it to generate text for a specific task. It combines the skills learned during training to generate search queries when necessary, based on the retrieval instructions provided in the prompt.
- Iterative Retrieval: When the LLM encounters a retrieval instruction in the prompt, it generates a search query and retrieves relevant documents from external sources. These documents are then used to augment the LLM’s knowledge and aid in generating the next part of the text.
- Dynamic Query Generation: The LLM dynamically generates search queries based on the context of the text it is generating and the retrieval instructions provided. This enables the LLM to adapt its retrieval behavior based on the specific information needs of the task at hand.
Working of Direct FLARE

Here’s how Direct FLARE works:
- Confidence-Based Active Retrieval: Direct FLARE operates on the principle of confidence-based active retrieval. At each step of text generation, the LLM first generates a temporary next sentence based on the context of the text generated so far. This temporary sentence is produced without conditioning on any retrieved documents.
- Query Formulation: The LLM then assesses the confidence of the temporary sentence it has generated. If the LLM is confident about the temporary sentence, it accepts it without retrieving additional information. However, if the LLM is uncertain about the temporary sentence, it formulates search queries based on the content of the temporary sentence.
- Dynamic Retrieval: Based on the formulated search queries, the LLM retrieves relevant documents from external sources. These documents contain information that may help the LLM resolve uncertainties or gain insights into the topic it is generating text about.
- Regeneration with Retrieved Information: After retrieving relevant documents, the LLM regenerates the next part of the text, incorporating insights gained from the retrieved information. This iterative process of generating, assessing confidence, formulating queries, retrieving documents, and regenerating text continues until the generation process is complete.
- Adaptive Retrieval Threshold: Direct FLARE uses a confidence threshold parameter to determine when to trigger retrieval. This threshold can be adjusted to control the frequency of retrieval during text generation, allowing for flexibility in the retrieval process.
Why Do We Need Cloud GPUs?
In this blog post, we will be using a cancer research PDF to implement FLARE with a Knowledge Graph and Llama 3 8B, which can greatly benefit from leveraging Cloud GPUs. These computational resources offer parallel processing capabilities, accelerating the training process and reducing the time required for model development and deployment. Cloud GPU instances can be dynamically scaled up or down based on workload requirements, ensuring efficient handling of varying data sizes and complexities inherent in large-scale medical datasets.
Cloud GPUs provide ample memory capacity, essential for processing large models and datasets without encountering memory constraints. Their optimized hardware configurations and software stack tailored for deep learning tasks lead to faster model convergence and improved performance. Despite incurring costs, cloud GPU instances offer a cost-effective solution compared to on-premises infrastructure, with pay-as-you-go pricing models ensuring efficient resource utilization.
Moreover, Cloud GPUs provide easy accessibility, which enables researchers to quickly access powerful computing resources without upfront investments or infrastructure management hassles. Given the parallel processing nature of FLARE models with Knowledge Graph integration, Cloud GPUs excel at accelerating graph traversal, entity linking, and relation extraction tasks, by enhancing the efficiency of medical research projects leveraging FLARE.
We will assume that you have access to advanced cloud GPUs from one of the providers: E2E Networks, AWS, Google Cloud, Azure or others. Open your Visual Studio code, and download the extension Remote Explorer and Remote SSH. Open a new terminal. Login to your local system with the following code:
ssh root@<your-ip-address>With this, you’ll be logged in to your node.
Implementing FLARE with Knowledge Graph
To get started with Forward-Looking Active RAG, we will first install all the required dependencies.
%pip install -q neo4j
%pip install -q llama-index-graph-stores-neo4j
%pip install -q llama-cpp-python
%pip install -q llama-index-llms-llama-cpp
%pip install -q llama-index-embeddings-huggingfaceWe’ll download the cancer research review PDF as our dataset.
!mkdir data
!wget https://www.illumina.com/content/dam/illumina-marketing/documents/products/research_reviews/cancer_research_review.pdf -P ./dataLet’s load the document using LlamaIndex SimpleDirectoryReader.
from llama_index.core import SimpleDirectoryReader
documents = SimpleDirectoryReader("./data").load_data()Then we’ll load the Hugging Face embeddings model.
from llama_index.embeddings.huggingface import HuggingFaceEmbedding
embed_model = HuggingFaceEmbedding(model_name="BAAI/bge-small-en-v1.5")After that, we’ll load the Llama 3 8B model from “The Bloke” from Hugging Face using Llama CPP.
from llama_index.llms.llama_cpp import LlamaCPP
from llama_index.core import Settings
model_url = "https://huggingface.co/QuantFactory/Meta-Llama-3-8B-Instruct-GGUF/blob/main/Meta-Llama-3-8B-Instruct.Q2_K.gguf"
llm = LlamaCPP(
model_url=model_url,
model_path=None,
temperature=0.1,
max_new_tokens=256,
context_window=3900,
generate_kwargs={},
model_kwargs={"n_gpu_layers": 1},
verbose=True,
)
Settings.llm = llm
Settings.embed_model = embed_model
Settings.chunk_size = 512After that, it is our turn to initiate the Neo4j Knowledge Graph. Simply visit the website and get started for free. Make an instance and download the file. This file will contain your username, password, and connection URI.
Set the variables.
username = "neo4j"
password = `your-password`
url = `your-connection-url`
database = "neo4j"Initiate the Neo4j Knowledge Graph.
from llama_index.core import KnowledgeGraphIndex
from llama_index.core import StorageContext
from llama_index.graph_stores.neo4j import Neo4jGraphStore
graph_store = Neo4jGraphStore(
username=username,
password=password,
url=url,
database=database,
)
storage_context = StorageContext.from_defaults(graph_store=graph_store)
index = KnowledgeGraphIndex.from_documents(
documents,
storage_context=storage_context,
max_triplets_per_chunk=2,
)Set the index query engine.
index_query_engine = index.as_query_engine(
include_text=True, response_mode="tree_summarize"
)As there are two types of FLARE, we’ll be implementing FLARE with Retrieval Instructions. For this framework, LlamaIndex has introduced FLAREInstructQueryEngine.
from llama_index.core.query_engine import FLAREInstructQueryEngine
flare_query_engine = FLAREInstructQueryEngine(
query_engine=index_query_engine,
max_iterations=7,
verbose=True,
)Now, it is time to pass the queries.
Question 1:
response = flare_query_engine.query(
"What is Gene Expression Analysis?"
)Query: What is Gene Expression Analysis?
Current response:
Lookahead response: [Search(What is Gene Expression Analysis?)]
Updated lookahead response: Gene expression analysis involves measuring the product of gene transcription, RNA processing, and epigenetic control. It provides an overview of the health of these processes and insights into molecular functions within the cell. Techniques like microarray-based mRNA analysis and sequencing-based mRNA analysis (mRNA-Seq) are commonly used in gene expression analysis, with mRNA-Seq being particularly advantageous due to its ability to detect modified RNAs, RNAs expressed at low levels, rapid changes in transcription, splice variants, fusion genes, and alternative polyadenylation sites.
Current response: Gene expression analysis involves measuring the product of gene transcription, RNA processing, and epigenetic control. It provides an overview of the health of these processes and insights into molecular functions within the cell. Techniques like microarray-based mRNA analysis and sequencing-based mRNA analysis (mRNA-Seq) are commonly used in gene expression analysis, with mRNA-Seq being particularly advantageous due to its ability to detect modified RNAs, RNAs expressed at low levels, rapid changes in transcription, splice variants, fusion genes, and alternative polyadenylation sites.
Lookahead response: [Search(What are some other techniques commonly used in gene expression analysis?)]
Updated lookahead response: Microarray-based mRNA analysis is a commonly used technique in gene expression analysis, providing valuable insights into molecular functions within the cell. Another technique that has advanced the field is sequencing-based mRNA analysis (mRNA-Seq), which offers the ability to detect modified RNAs, RNAs expressed at low levels, rapid changes in transcription, splice variants, fusion genes, and alternative polyadenylation sites.
Current response: Gene expression analysis involves measuring the product of gene transcription, RNA processing, and epigenetic control. It provides an overview of the health of these processes and insights into molecular functions within the cell. Techniques like microarray-based mRNA analysis and sequencing-based mRNA analysis (mRNA-Seq) are commonly used in gene expression analysis, with mRNA-Seq being particularly advantageous due to its ability to detect modified RNAs, RNAs expressed at low levels, rapid changes in transcription, splice variants, fusion genes, and alternative polyadenylation sites. Microarray-based mRNA analysis is a commonly used technique in gene expression analysis, providing valuable insights into molecular functions within the cell. Another technique that has advanced the field is sequencing-based mRNA analysis (mRNA-Seq), which offers the ability to detect modified RNAs, RNAs expressed at low levels, rapid changes in transcription, splice variants, fusion genes, and alternative polyadenylation sites.
Lookahead response: Gene expression analysis is crucial for understanding the health of gene transcription, RNA processing, and epigenetic control within cells. It utilizes techniques like microarray-based mRNA analysis and sequencing-based mRNA analysis (mRNA-Seq) to provide insights into molecular functions and detect various RNA modifications and expression levels.
...
...
...
Updated lookahead response: Some applications of gene expression analysis in cancer research include detecting driver mutations, confirming fusion genes, assessing the impact of copy number variations (CNVs) on expression levels, and confirming gene mutations through mRNA sequencing.
Current response: Gene expression analysis involves measuring the product of gene transcription, RNA processing, and epigenetic control. It provides an overview of the health of these processes and insights into molecular functions within the cell. Techniques like microarray-based mRNA analysis and sequencing-based mRNA analysis (mRNA-Seq) are commonly used in gene expression analysis, with mRNA-Seq being particularly advantageous due to its ability to detect modified RNAs, RNAs expressed at low levels, rapid changes in transcription, splice variants, fusion genes, and alternative polyadenylation sites. Microarray-based mRNA analysis is a commonly used technique in gene expression analysis, providing valuable insights into molecular functions within the cell. Another technique that has advanced the field is sequencing-based mRNA analysis (mRNA-Seq), which offers the ability to detect modified RNAs, RNAs expressed at low levels, rapid changes in transcription, splice variants, fusion genes, and alternative polyadenylation sites.
Lookahead response: [Search(What are some applications of gene expression analysis in cancer research?)]
Updated lookahead response: Some applications of gene expression analysis in cancer research include detecting driver mutations, confirming fusion genes, assessing the impact of copy number variations (CNVs) on expression levels, and confirming gene mutations through mRNA sequencing.Answer:
print(response)Gene expression analysis involves measuring the product of gene transcription, RNA processing, and epigenetic control. It provides an overview of the health of these processes and insights into molecular functions within the cell.
Techniques like microarray-based mRNA analysis and sequencing-based mRNA analysis (mRNA-Seq) are commonly used in gene expression analysis, with mRNA-Seq being particularly advantageous due to its ability to detect modified RNAs, RNAs expressed at low levels, rapid changes in transcription, splice variants, fusion genes, and alternative polyadenylation sites. Microarray-based mRNA analysis is a commonly used technique in gene expression analysis, providing valuable insights into molecular functions within the cell. Another technique that has advanced the field is sequencing-based mRNA analysis (mRNA-Seq), which offers the ability to detect modified RNAs, RNAs expressed at low levels, rapid changes in transcription, splice variants, fusion genes, and alternative polyadenylation sites.Question 2:
response = flare_query_engine.query(
"What is RNA Editing?"
)Query: What is RNA Editing?
Current response:
Lookahead response: [Search(What is RNA Editing?)]
Updated lookahead response: RNA editing involves the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs). This process is recognized by the splicing and translational machineries as a guanosine.
Current response: RNA editing involves the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs). This process is recognized by the splicing and translational machineries as a guanosine.
Lookahead response: [Search(What is the significance of adenosine to inosine conversion in RNA editing?)]
Updated lookahead response: The significance of adenosine to inosine conversion in RNA editing lies in the fact that this process is the most frequent type of RNA editing in humans. Adenosine deaminases acting on RNA (ADARs) catalyze this conversion, where adenosine is changed to inosine. Subsequently, the splicing and translational machineries recognize inosine as guanosine. This conversion plays a crucial role in altering the genetic information encoded in RNA, impacting gene expression and potentially contributing to various biological processes and diseases.
Current response: RNA editing involves the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs). This process is recognized by the splicing and translational machineries as a guanosine. The significance of adenosine to inosine conversion in RNA editing lies in the fact that this process is the most frequent type of RNA editing in humans. Adenosine deaminases acting on RNA (ADARs) catalyze this conversion, where adenosine is changed to inosine. Subsequently, the splicing and translational machineries recognize inosine as guanosine. This conversion plays a crucial role in altering the genetic information encoded in RNA, impacting gene expression and potentially contributing to various biological processes and diseases.
Lookahead response: [Search(What are the biological processes impacted by RNA editing?)]
Updated lookahead response: The biological processes impacted by RNA editing include the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs), which is recognized as guanosine by the splicing and translational machineries. Additionally, some tumor genomes exhibit a higher percentage of RNA-DNA differences compared to their matched normal genomes.
...
...
...
Current response: RNA editing involves the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs). This process is recognized by the splicing and translational machineries as a guanosine. The significance of adenosine to inosine conversion in RNA editing lies in the fact that this process is the most frequent type of RNA editing in humans. Adenosine deaminases acting on RNA (ADARs) catalyze this conversion, where adenosine is changed to inosine. Subsequently, the splicing and translational machineries recognize inosine as guanosine. This conversion plays a crucial role in altering the genetic information encoded in RNA, impacting gene expression and potentially contributing to various biological processes and diseases. The biological processes impacted by RNA editing include the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs), which is recognized as guanosine by the splicing and translational machineries. Additionally, some tumor genomes exhibit a higher percentage of RNA-DNA differences compared to their matched normal genomes. RNA editing can impact gene expression and contribute to various biological processes and diseases. Examples of biological processes impacted by RNA editing include the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs), which is recognized by the splicing and translational machineries as a guanosine. Additionally, some tumor genomes exhibit a higher percentage of RNA-DNA differences compared to their matched normal genomes. RNA editing can impact gene expression and contribute to various biological processes and diseases. Examples of biological processes impacted by RNA editing include the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs), which is recognized by the splicing and translational machineries as a guanosine.
Lookahead response: The conversion of adenosine to inosine by ADARs is crucial in altering genetic information encoded in RNA, impacting gene expression and potentially contributing to various biological processes and diseases.
Updated lookahead response: The conversion of adenosine to inosine by ADARs is crucial in altering genetic information encoded in RNA, impacting gene expression and potentially contributing to various biological processes and diseases.Answer:
print(response)RNA editing involves the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs).
This process is recognized by the splicing and translational machineries as a guanosine. The significance of adenosine to inosine conversion in RNA editing lies in the fact that this process is the most frequent type of RNA editing in humans.
Adenosine deaminases acting on RNA (ADARs) catalyze this conversion, where adenosine is changed to inosine. Subsequently, the splicing and translational machineries recognize inosine as guanosine.
This conversion plays a crucial role in altering the genetic information encoded in RNA, impacting gene expression and potentially contributing to various biological processes and diseases.
The biological processes impacted by RNA editing include the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs), which is recognized as guanosine by the splicing and translational machineries. Additionally, some tumor genomes exhibit a higher percentage of RNA-DNA differences compared to their matched normal genomes. RNA editing can impact gene expression and contribute to various biological processes and diseases.
Examples of biological processes impacted by RNA editing include the conversion of adenosine to inosine by adenosine deaminases acting on RNA (ADARs), which is recognized by the splicing and translational machineries as a guanosine.Conclusion
To conclude, it was easy to implement Forward-Looking Active RAG using Neo4j Knowledge Graph and Llama 3 8B GGUF model on the LlamaIndex framework. We experimented with FLARE with the Retrieval Instructions method, and the results were great.
References
https://arxiv.org/pdf/2305.06983.pdf
https://docs.llamaindex.ai/en/stable/examples/query_engine/flare_query_engine.html



-Feature-Image.CjBFdO7y_Z1bTNvg.webp)